Electrocatalytic Hydrogenation
Effect of electrode potential, electrolyte species, and co-reactants on electrocatalytic hydrogenation of organics using DFT and AIMD.
VASPJDFTxASE
Computational Catalysis · ML Potentials · Agentic AI
PhD Candidate · Chemical Engineering
University of Michigan, Ann Arbor

Interfacial electrochemistry, multiscale modeling, and scientific machine learning for materials and catalysis.
I am a PhD candidate in Chemical Engineering and Scientific Computing at the University of Michigan. I focus on atomistic modeling for materials science and catalysis, combining advanced computational methods to understand and predict complex interfacial phenomena. My work integrates density functional theory (DFT), ab initio molecular dynamics (AIMD), classical molecular dynamics, machine-learning interatomic potentials, and emerging agentic AI and physics-informed ML architectures. My goal is to improve predictive accuracy, enable materials discovery, and provide mechanistic explanations for experimental observations.
My doctoral research centers on the electrolyte–electrode interface, investigating how electrolyte composition and co-reactants influence adsorption, interfacial structure, and reaction mechanisms in electrochemical systems. I develop and benchmark MLIP models using MACE, DeepMD, and GRACE architectures to study water adsorption isotherms on different metals, and I run large-scale molecular dynamics simulations that couple DFT with machine-learning acceleration. I also integrate these approaches with analytical models to connect computation with experiment. I am also interested in agentic AI workflows for autonomous discovery in quantum chemistry, combining multi-agent planning with DFT-based mechanistic modeling and developing goal-evolving agents for scientific discovery.
I am motivated by bridging cutting-edge computational science with sustainable energy challenges. Through interdisciplinary collaboration, I aim to contribute to impactful research while promoting inclusivity, innovation, and environmental sustainability.
Atomistic modeling, electrocatalysis, and AI-driven discovery.
Effect of electrode potential, electrolyte species, and co-reactants on electrocatalytic hydrogenation of organics using DFT and AIMD.
Multi-agent LLM workflows for autonomous transition-state discovery, combining planning with DFT-based mechanistic modeling.
Estimating potential-dependent physicochemical properties at metal–electrolyte interfaces using MACE architecture at Lawrence Livermore National Lab.
Role of aqueous ions on adsorption of aromatic organics on Ag surfaces and synergistic effects in organic mixtures.
Conversion of lignocellulosic biomass and agricultural residues into bio-oil at the University of Alberta.
Scalable HPC pipelines for DFT and MD workflows, Slurm/PBS orchestration, and GPU-accelerated computing.
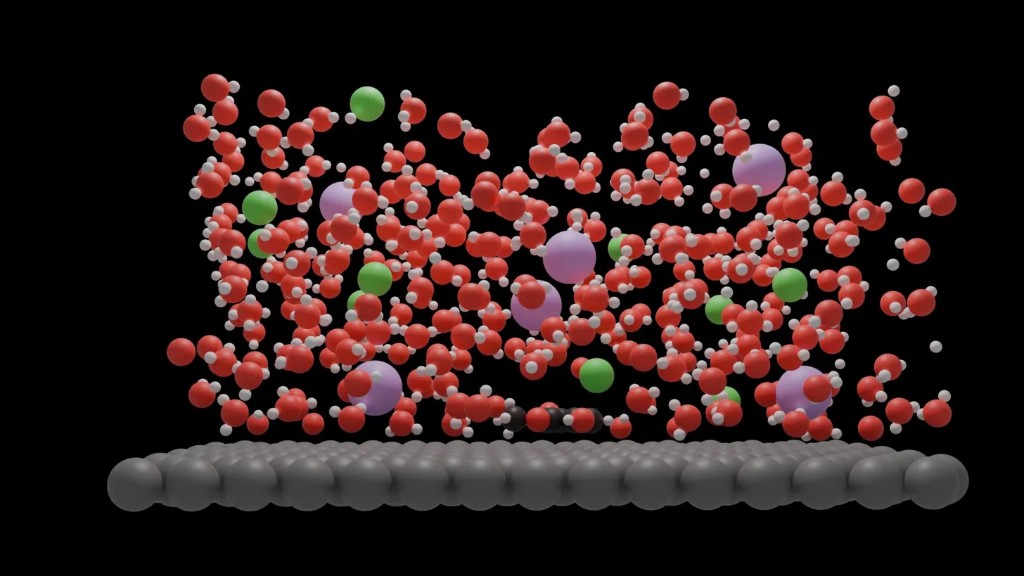
Molecular Dynamics Simulation — Metal–electrolyte interface showing water molecules, co-ions, and adsorbed organics at a platinum surface.
Peer-reviewed articles and preprints (newest first). Each entry includes a graphical abstract; titles and thumbnails link to the publisher, arXiv, or repository.
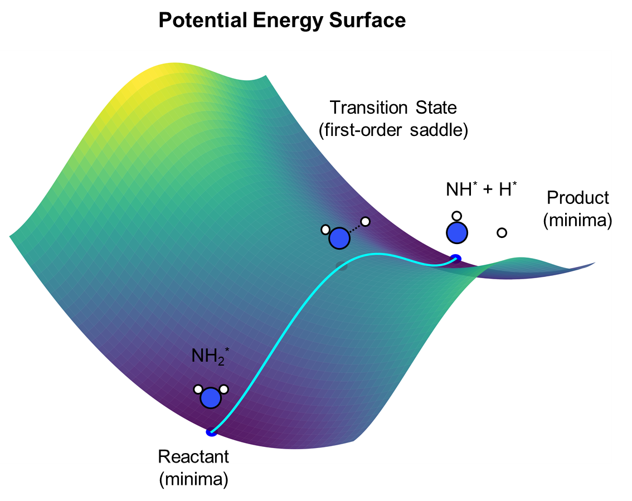


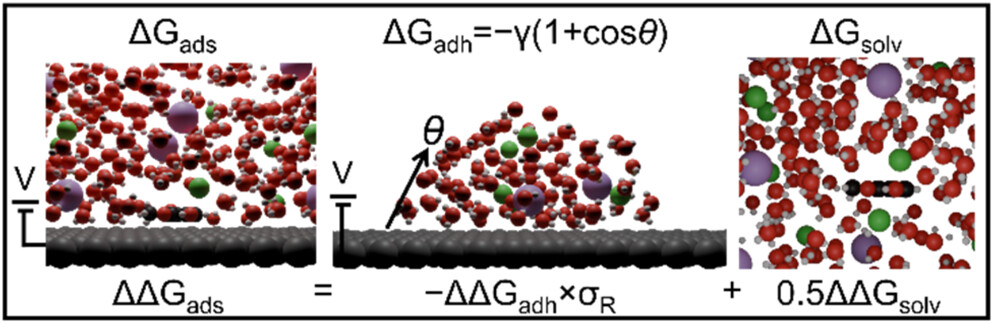
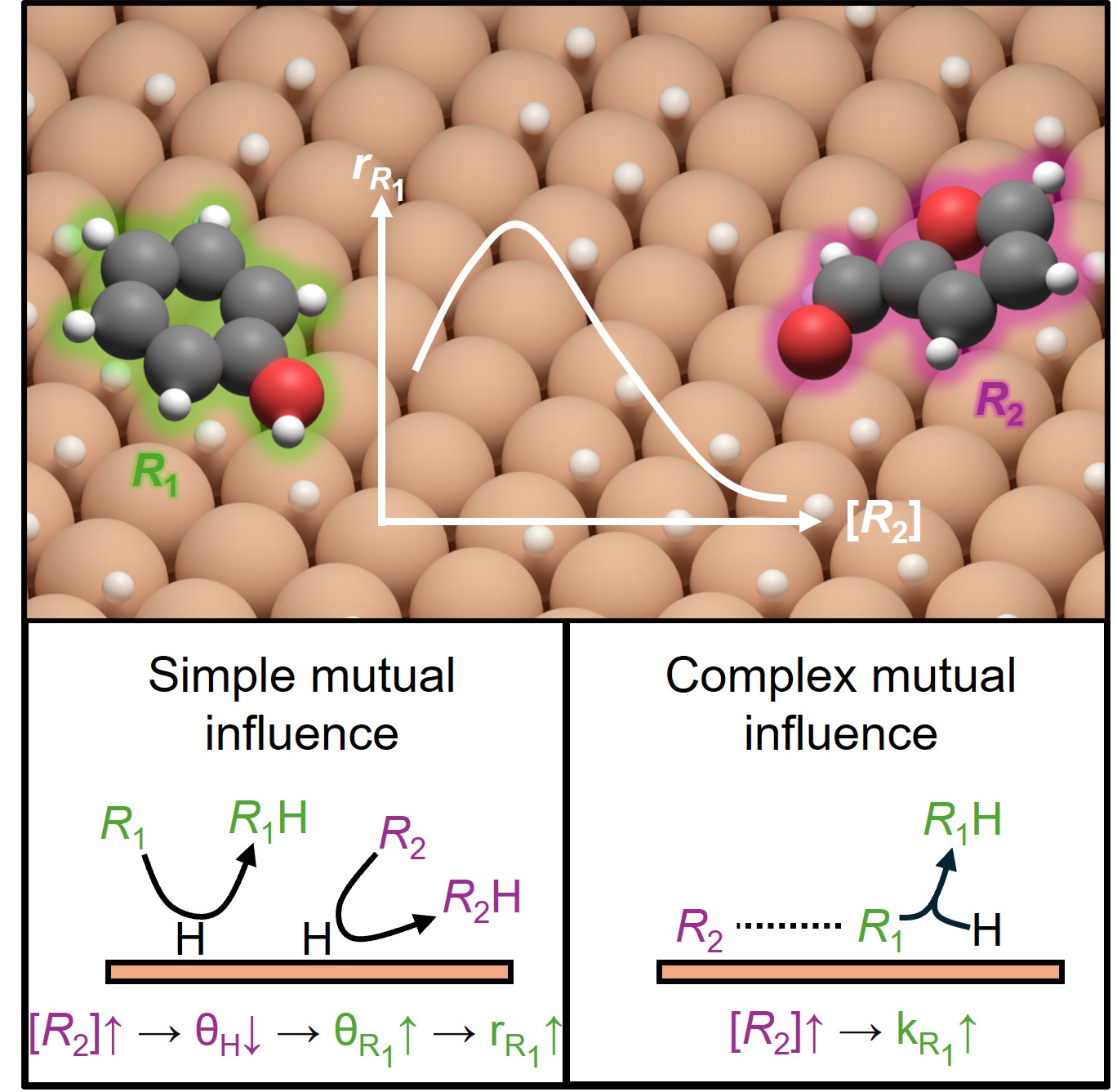
Tools and methods across modeling, ML, and computing.
Recognition for research, travel, and innovation.
Top 25 teams globally; agentic AI for materials science.
For exceptional advancements in catalysis.
University of Michigan (2022, 2023, 2024).
University of Alberta; top 5% incoming cohort.
University of Alberta; extraordinary research.
Indian Academy of Sciences; top 10% applicants.